
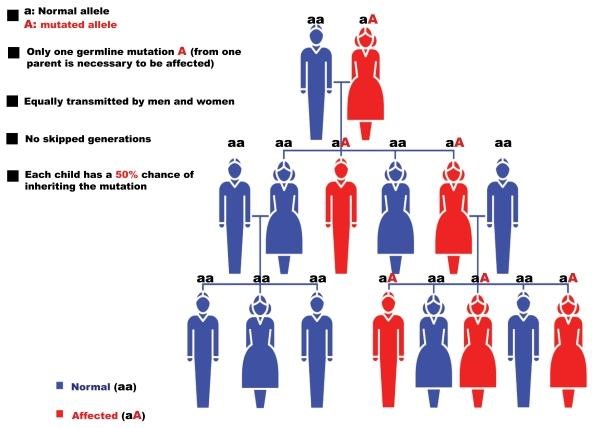
Autosomal-dominanter Erbgang
Der Erbgang bei ARVC / ACM ist in den meisten Fällen autosomal-dominant. Das heißt: wer eine Genveränderung aufweist, vererbt sie unabhängig vom Geschlecht mit einer Wahrscheinlichkeit von 50% an seine Nachkommen. Im Umkehrschluss hat also jedes Kind eines Genvariantenträgers eine Chance von 50%, die Variante NICHT in sich zu tragen.
Variantenträger oder Anlageträger werden also Personen genannt, die eine Genvariante für ARVC in sich tragen. Diese Personen können im Laufe ihres Lebens an ARVC erkranken, dies muss jedoch nicht zwingend geschehen und ist von vielen Einflussfaktoren abhängig. Das nennt man im Medizinischen unvollständige oder inkomplette Penetranz: nur ein gewisser Prozentsatz der Genvariantenträger erkrankt auch tatsächlich. Diese Prozentzahl schwankt erheblich und unterscheidet sich je nach betroffenem Gen.
Generationen werden beim autosomal-dominanten Erbgang nicht übersprungen. Wenn ein Nachkomme die Genvariante nicht geerbt hat, dann können weder er noch seine eigenen Nachkommen erkranken oder die Anlage für die Erkrankung weitervererben. In diesem Familienzweig ist die ARVC dann „ausgestorben“.
Ferrari, S. et al, 2011, Retinitis Pigmentosa: Genes and Disease Mechanisms, 12:238-49
Autosomal rezessiver Erbgang
Nur selten ist der Erbgang autosomal-rezessiv, beispielsweise bei der Naxos-Krankheit (JUP-Genvariante) oder dem Carvajal Syndrom (DSP-Genvariante). Diese Erkrankungen kommen aber in Deutschland nur sehr selten vor. Bei einem rezessiven Erbgang müssen zwei Kopien des veränderten Gens (von Mutter und Vater) bei einer Person zusammenkommen, um die Erkrankung zu verursachen.
In der nächsten Generation gibt es dann 25% erkrankte Personen mit zwei veränderten Kopien des Gens, 25% gesunde Personen die keine Variante geerbt haben, und 50% Anlageträger, die selbst nicht erkranken, aber eine Kopie der Variante in sich tragen, die sie weitervererben können. Bei einem rezessivem Erbgang können also Generationen übersprungen werden, die Erkrankung taucht nicht in jeder Generation auf.
- WICHTIG
Die meisten Betroffenen in Deutschland, die eine DSP- oder JUP-Varianten aufweisen, haben nicht die Naxos- oder Carvajal-Erkrankung, sondern eine autosomal-dominant vererbte Form der arrhythmogenen Kardiomyopathie, die neben der typischen rechtsbetonten Form (ARVC) auch linksbetont (ALVC, NDLVC) oder beidseitig (ACM) ausgeprägt sein kann.
Mehr erfahren
Genetik-Begriffe erklärt
Medizinische Abkürzungen
Medizinische Fachbegriffe
Fachartikel Genetik
Vortragsfolien „Genetik bei ARVC, molekulare Autopsie und Register“, Prof. Dr. rer. nat. Hendrik Milting, Prof. Dr. Peter van Tintelen (2019)
Vortragsfolien „Genetik und Möglichkeiten der Präimplantationsdiagnostik“, Dr. Teresa Neuhann (2016)
Video des Vortrags „ Genetik bei ARVC“, Prof. Dr. med. Eric Schulze-Bahr (2023)
Video des Vortrags „Genetik bei ARVC, molekulare Autopsie und Register“, Prof. Dr. rer. nat. Hendrik Milting, Prof. Dr. Peter van Tintelen (2019)
FAQ Erbgang – Häufig gestellte Fragen
Bei autosomal-dominantem Erbgang ist das 50%. Trotzdem gibt es Familien, bei denen alle Kinder und Familien, bei denen kein Kind betroffen ist. Die Wahrscheinlichkeit ist für jedes einzelne Kind 50%, und die Wahrscheinlichkeit für das zweite sinkt nicht, wenn bereits eines betroffen ist.
Bei ARVC ist das tatsächlich in über 98% der Fall, selbst wenn die Eltern keinerlei Symptome haben. Bei ARVC gibt es nur zu ca. 1,3% Neumutationen, die bei einem Individuum neu entstanden sind und nicht von einem Elternteil vererbt wurden.
In einigen wenigen Familien tragen sogar beide Eltern je eine (oder mehrere) Genvarianten und vererben diese in wechselnden Konstellationen an ihre Nachkommen.
Quelle:
Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo
van Lint FHM, Murray B, Tichnell C, et al. Circ Genom Precis Med. 2019 Aug;12(8):e002467
https://doi.org/10.1161/circgen.119.002467
Gendiagnostik bei kardiovaskulären Erkrankungen – Konsensuspapier der Deutschen Gesellschaft für Kardiologie (DGK), der Gesellschaft für Humangenetik (GfH) und der Deutschen Gesellschaft für Pädiatrische Kardiologie (DGPK)
Schulze-Bahr E, Klaassen S, Gerull B et al., Kardiologie 2023; 17:300–349
https://doi.org/10.1007/s12181-023-00622-3
Pocket-Leitlinie: Kardiomyopathien - Leitlinien für das Management von Kardiomyopathien (Version 2023)
Meder B, Eckardt L, Falk V et al. Deutsche Gesellschaft für Kardiologie – Herz- und Kreislaufforschung e.V. (2023) - ESC Pocket Guidelines, Börm Bruckmeier Verlag GmbH
https://leitlinien.dgk.org/2024/pocket-leitlinien-kardiomyopathien-version-2023/
2023 ESC Guidelines for the management of cardiomyopathies
Arbelo E, Protonotarios A, Gimeno JR et al. ESC Scientific Document Group, Eur Heart J. 2023 Aug 25:ehad194
https://doi.org/10.1093/eurheartj/ehad194
Vortrag „Genetik bei ARVC“
Prof. Dr. med. Eric Schulze-Bahr, Institut für Genetik von Herzerkrankungen, Universitätsklinikum Münster
Symposium "ARVC-Selbsthilfe trifft Fachwissen", München, 17.06.2023
https://www.youtube.com/watch?v=XprLLxfLHso
Vortrag „ACM: Kausale Gene, Umgang mit Varianten unklarer Signifikanz (VUS) und genetische Diagnostik bei Kindern“
PD Dr. med. Dominik Westphal, Munich Consortium des European Reference Networks ERN GUARD-Heart, Klinikum Rechts der Isar der TU München
Symposium "ARVC-Selbsthilfe trifft Fachwissen", München, 16.06.2023
https://www.youtube.com/watch?v=XprLLxfLHso
Pocket-Leitlinie: Ventrikuläre Arrhythmien und Prävention des plötzlichen Herztodes (Version 2022)
Eckardt L, Bosch R, Falk V, et al. Deutsche Gesellschaft für Kardiologie – Herz- und Kreislaufforschung e.V. (2023) - ESC Pocket Guidelines, Börm Bruckmeier Verlag GmbH
https://leitlinien.dgk.org/2023/pocket-leitlinie-ventrikulaere-arrhythmien-und-praevention-des-ploetzlichen-herztodes-version-2022/
2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death
Zeppenfeld K, Tfelt-Hansen J, de Riva M et al. Eur Heart J. 2022 Oct 21;43(40):3997-4126
https://doi.org/10.1093/eurheartj/ehac262
European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases
Wilde AAM, Semsarian C, Márquez MF et al. Heart Rhythm. 2022 Jul;19(7):e1-e60
https://doi.org/10.1016/j.hrthm.2022.03.1225
2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy
Towbin JA, McKenna WJ, Abrams DJ et al. Heart Rhythm Volume 16, ISSUE 11, e301-e372, November 01, 2019
https://doi.org/10.1016/j.hrthm.2019.05.007
Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo
van Lint FHM, Murray B, Tichnell C, et al. Circ Genom Precis Med. 2019 Aug;12(8):e002467
https://doi.org/10.1161/circgen.119.002467
Vortrag „Genetik bei ARVC, molekulare Autopsie und Register“
Prof. Dr. rer. nat. Hendrik Milting, Kardiogenetik Herz- und Diabeteszentrum Bad Oeynhausen
Prof. Dr. Peter van Tintelen, Kardiogenetik, UMC Utrecht, Niederlande
Symposium "ARVC-Selbsthilfe trifft Fachwissen", München, 23.02.2019
Vortragsfolien
https://www.youtube.com/watch?v=PzkIJuaihoQ
Arrhythmogenic Right Ventricular Cardiomyopathy: An Update on Pathophysiology, Genetics, Diagnosis, and Risk Stratification
Paul M, Wichter T, Fabritz L et al. Herzschrittmacherther Elektrophysiol. 2012 Sep;23(3):186-95
https://doi.org/10.1007/s00399-012-0233-7
Die arrhythmogene rechtsventrikuläre Dysplasie/Kardiomyopathie
Saguner A, Brunckhorst C, Duru F. Cardiovascular Medicine 2011; 14 (11): 303-314
https://cardiovascmed.ch/article/doi/cvm.2011.01623
Letzte Aktualisierung: 01.10.2024
Fachliche Überprüfung: Prof. Dr. med. Daniela Husser-Bollmann
Die Informationen dieser Website ersetzen keine ärztliche Beratung. Jeder Fall ist individuell. Gesamteinschätzung, Diagnose und Auswahl einer angemessenen Therapie gehören daher immer in die Hände eines erfahrenen Arztes. Gerne helfen wir, Kontakte zu Experten herzustellen, insbesondere auch, wenn es Probleme mit der genetischen Diagnostik gibt.